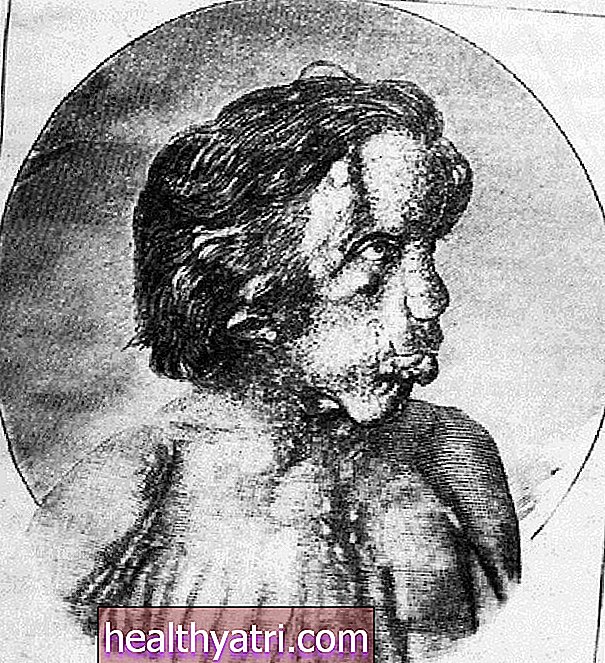
Smith Lemli Opitzův syndrom je vrozená vývojová porucha charakterizovaná kromě jiných projevů charakteristickými rysy obličeje, intelektuálními poruchami a poruchami učení, problémy s chováním a malou hlavou (mikrocefalií). Vedle malformací důležitých orgánů, jako jsou ledviny, srdce, genitálie a střevní trakt, vykazují děti s tímto stavem charakteristiky autismu a poruchy pozornosti s hyperaktivitou (ADHD).Většina pacientů s tímto onemocněním má spojený druhý a třetí prst a některé mohou mít další prsty. Tento stav je relativně vzácný a postihuje přibližně jednoho z každých 20 000 až 60 000 kojenců.
SeventyFour / Getty ImagesPříznaky
Známky syndromu Smitha Lemliho Opitze se vyskytují při narození a jejich závažnost se velmi liší. V 80 procentech až 99 procentech těchto případů. jsou vidět tyto vlastnosti:
- Webbed toes: Common feature of the condition is fusing between the second and third toes, a condition called „syndactyl."
- Mentální postižení: I když se míra může lišit, tato podmínka často vede k poruchám učení.
- Abnormálně malá lebka: Menší než průměrná velikost lebky, stav zvaný mikrocefalie, je dalším charakteristickým znakem.
- Abnormální rysy obličeje: Pacienti se syndromem Smith Lemli Opitz mají charakteristické rysy obličeje včetně menší dolní čelisti a širokého plochého nosu. Ve vzácnějších případech mohou mít jedinci klesající víčka, kočičí oči, malé nebo chybějící oči a také široká ústa.
- Obtížné krmení: U kojenců může tento stav vést k obtížnému kojení a ovlivnit tak vývoj.
- Nižší svalový tonus: Společnou charakteristikou syndromu je nižší než průměrný svalový tonus.
Existuje mnoho méně častých příznaků, které se vyskytují kdekoli od 5 do 79 procent případů, včetně:
- Abnormality zubního vývoje: Včasná erupce dospělých zubů a zvětšení dásní jsou příznaky syndromu Smith Lemli Opitz.
- Nejednoznačné genitálie: Genitálie postižených mohou být méně definované. U mužů je větší pravděpodobnost, že to zažijí, s nedostatečně vyvinutým penisem a nesestouplými varlaty.
- Porucha hyperaktivity s deficitem pozornosti (ADHD): Tato vývojová porucha je charakterizována obtížemi regulovat chování a impulsy i hyperaktivitou.
- Autismus: Tento stav, známý také jako porucha autistického spektra (ASD), vede ke zhoršení sociálních dovedností, řeči a neverbálních komunikačních schopností a také k opakovanému chování.
- Srdeční vady: Srdeční vady spojené se syndromem Smith Lemli Opitz zahrnují vznik díry ve stěně mezi dvěma horními komorami (defekt síňového septa) nebo jednoho mezi dolními komorami (defekt komorového septa).
- Změněná anatomie ruky: Pacienti s tímto onemocněním mohou mít extra malé prsty na rukou a nohou. Poloha palce může být navíc atypická tím, že je blíže zápěstí. Byly také hlášeny plovací blány. Rovněž byla hlášena drápová ruka, atypické zakřivení prstů.
- Fotocitlivost: V mnoha případech je pokožka postižených mimořádně citlivá na sluneční světlo.
- Častá infekce: Lidé se syndromem mají zvýšené riziko bakteriální infekce.
- Rozštěp jazyka: V přibližně pěti až 30 procentech případů budou mít postižení rozštěp jazyka, ve kterém je špička rozdělena.
- Abnormality v páteři: Spolu s dalšími deformacemi obratlů může tento stav doprovázet skolióza - boční zakřivení páteře - stejně jako kyfóza nebo hrb.
- Záchvaty: Lidé s tímto onemocněním jsou náchylnější k rozvoji záchvatů.
- Nedobrovolné pohyby očí: Syndrom může doprovázet také nekontrolované a rychlé pohyby očí (nystagmus).
Příčiny
Smith Lemli Opitzův syndrom je genetická porucha způsobená mutací genu DHCR7. Tento gen reguluje důležitý enzym, 7-dehydrocholesterol reduktázu, který se podílí na produkci cholesterolu v těle. Mezi svými funkcemi je cholesterol hlavní složkou buněčných membrán a pomáhá vytvářet myelin, látku, která chrání mozkové buňky (neurony). Hraje také významnou roli při správném trávení.
Mutace DHCR7 způsobuje nedostatek 7-dehydrocholesterol reduktázy, což způsobuje deficity v produkci cholesterolu. Umožňuje také hromadění toxických vedlejších produktů cholesterolu v těle, což brání vývoji a růstu v různých tělesných systémech. Přesný mechanismus, jak tento nedostatek cholesterolu vede k syndromu Smith Lemli Opitz, je stále zkoumán.
Tento genetický defekt sleduje tento stav, který se nazývá „autozomálně recesivní vzor“, což znamená, že k jeho vývoji jsou nezbytné obě kopie genu - jedna od každého rodiče. To znamená, že rodiče těch, kteří mají tento stav, nesou gen, ale nemusí nutně mít samotné příznaky.
Diagnóza
Stejně jako u jiných vrozených onemocnění zahrnuje diagnóza Smitha Lemli Opitze hodnocení fyzických příznaků a testování poměru 7-dehydrocholesterol reduktázy k cholesterolu. To se provádí pomocí krevních testů podezřelých případů. Kromě toho může prenatální genetické testování také detekovat mutace genu DHCR7, které vedou k rozvoji stavu.
Léčba
Přijetí této podmínky vyžaduje koordinované úsilí; protože na tento stav neexistuje přímý lék, je třeba účinně zvládat příznaky a projevy. Mezi tyto přístupy patří:
- Suplementace cholesterolu: I když je zapotřebí dalšího výzkumu k posouzení účinnosti tohoto přístupu, dieta bohatá na cholesterol - vedle užívání doplňků - může pomoci snížit některé příznaky.
- Fyzikální terapie: Přístupy fyzikální a pracovní terapie, pokud jsou poskytovány včas, mohou pomoci s postižením spojeným se stavem.
- Lékařské ošetření: K dispozici jsou přístupy k řešení některých fyzických příznaků syndromu Smith Lemli Opitz, včetně zažívacích potíží, zrakových problémů, stejně jako obličejových a jiných deformit.
- Dohled: Úspěšné zvládnutí tohoto stavu vyžaduje důsledné sledování fyzických příznaků, zpoždění vývoje a dietních faktorů.
Prognóza
Dobrou zprávou je, že pokud bude syndrom Smith Lemli Opitz řádně spravován a bude poskytována adekvátní lékařská péče, u osob s tímto onemocněním bude mít normální průměrnou délku života. Nezávislý život je nepravděpodobný kvůli těžkému mentálnímu postižení, které často doprovází tento syndrom. Je pozoruhodně vážně ovlivněno přežití kojenců se závažnými příznaky a existuje šance na smrt během několika měsíců.
Zvládání
Hlavní vrozená porucha, jako je syndrom Smith Lemli Opitz, představuje významnou výzvu pro postiženou osobu, její rodinu i lékaře. I když je možné úspěšné řízení, není pochyb o tom, že z tohoto břemene vyplývají významné psychologické spády. Ti, kteří se nacházejí v pozici péče o někoho s tímto onemocněním, mohou považovat za užitečné poradenské skupiny nebo skupiny podporující postižení. Zejména zdroje, jako jsou odkazy na nejnovější služby výzkumu a podpory, shromažďuje nadace Smith Lemli Opitz / RSH Foundation.
Slovo od Verywell
Stav, který je tento vysilující a obtížný a může ovlivnit tolik aspektů kvality života, se může zdát zdrcující. To znamená, že nejen že se stávající léčebné přístupy k syndromu Smith Lemli Opitz neustále zdokonalují a zdokonalují, ale výzkum této poruchy stále probíhá. Jakmile se lékařská komunita dozví více o příčinách a následcích tohoto stavu - a také o účinnosti přístupů k léčbě - prognóza a kvalita života postižených se pouze zlepší.