Apertův syndrom je genetický stav, který každoročně postihuje přibližně jednoho z 65 000 až 88 000 novorozenců. Mezi běžné rysy u lidí s Apertovým syndromem patří mimo jiné předčasně fúzované kosti lebky, fúze některých prstů na rukou a nohou. Ačkoli tento stav způsobuje různé stupně tělesného i duševního postižení, lidé s Apertovým syndromem mohou růst, aby si užívali plný a produktivní život.
Pokud vy nebo někdo, koho znáte, čeká dítě s Apertovým syndromem nebo se jen chcete dozvědět něco více o tomto stavu, je vždy užitečné vybavit se znalostmi o příznacích, příčinách, diagnostice, léčbě a zvládání.
gorodenkoff / Getty ImagesPříznaky
Děti s Apertovým syndromem mají určité kosti lebky, které se předčasně spojují v děloze (před narozením) a způsobují stav známý jako kraniosynostóza. Tato časná fúze zabraňuje růstu lebky tak, jak by měla, a ovlivňuje tvar hlavy a obličeje. Mezi běžné rysy obličeje u lidí s Apertovým syndromem patří:
- Vpadlý vzhled obličeje
- Vypouklé a / nebo široce posazené oči
- Zobákový nos
- Nedostatečně vyvinutá horní čelist
- Přeplněné zuby a další problémy se zuby
Protože časná fúze lebky může způsobit problémy vyvíjejícímu se mozku, mohou mít lidé s Apertovým syndromem také kognitivní postižení. Rozsah zpoždění vývoje a mentálního postižení se velmi liší - může být kdekoli od normálního po střední.
Mezi další charakteristiky a stavy, které se mohou vyskytnout u lidí s Apertovým syndromem, patří:
- Syndactyly (minimálně tři prsty na každé ruce a noze, které mohou být spojeny nebo spojeny)
- Polydaktylie (méně časté, ale na rukou nebo nohou mohou být další číslice)
- Ztráta sluchu
- Nadměrné pocení (hyperhidróza)
- Nadměrně mastná pleť a silné akné
- Záplaty chybějících vlasů v obočí
- Rozštěp patra
- Opakované infekce uší
- Tavené kosti na krku (krční obratle)
- Otvory ve stěně srdeční komory
- Blokování jícnu
- Špatně umístěný konečník
- Blokování pochvy
- Kryptorchismus (neschopnost varlat sestoupit do šourkového vaku)
- Rozšíření ledvin v důsledku zablokování toku moči
Příčiny
Apertův syndrom je způsoben mutací genu FGFR2. Podle National Institutes of Health Spojených států „Tento gen produkuje protein nazývaný receptor fibroblastového růstového faktoru 2. Mezi jeho více funkcemi tento protein signalizuje nezralým buňkám, aby se staly kostí buňky během embryonálního vývoje. Mutace v určité části DNAFGFR2 Gen mění protein a způsobuje prodlouženou signalizaci, která může podporovat předčasné spojení kostí v lebce, rukou a nohou. “
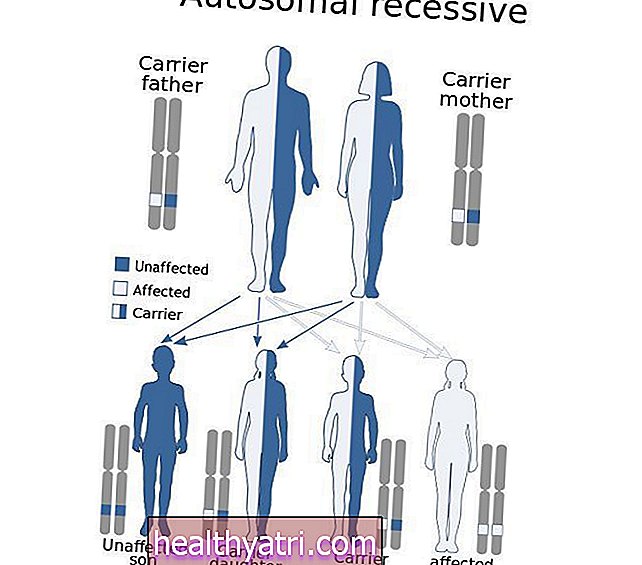
Ačkoli je tento stav genetický, téměř vždy se vyskytuje u lidí bez rodinné anamnézy Apertova syndromu, což znamená, že je způsoben novou mutací.
Lidé s Apertovým syndromem však mohou předat geny svým dětem. Pokud k tomu dojde, nemoc se přenáší jako autozomálně dominantní porucha.
Diagnóza
Lékaři mohou mít podezření na Apertův syndrom před narozením kvůli abnormálnímu vývoji lebky. Oficiální diagnóza je stanovena genetickým vyšetřením, které se provádí krevním testem. Toho lze dosáhnout pomocí amniocentézy, zatímco je matka ještě těhotná, pokud existuje podezření na Apertův syndrom.
Často se identifikuje na ultrazvuku kvůli abnormálně se vyvíjejícím kostem lebky. Fetální MRI však může poskytnout mnohem více podrobností o mozku než ultrazvuk. Potvrzení diagnózy se provádí krevními testy na gen, který ji způsobuje.
Léčba
Neexistuje žádná léčba, která by „vyléčila“ Apertův syndrom, protože se jedná o genetickou poruchu. Existuje však mnoho terapií, operací a dalších intervencí, které mohou zlepšit kvalitu života osoby s Apertovým syndromem. Potřebné konkrétní zásahy budou záviset na jednotlivci a na tom, jak jsou ovlivněny.
Mezi běžné operace u dětí s Apertovým syndromem patří:
- Lebka přetváření
- Frontální-orbitální pokrok (pro zvětšení prostoru v čele a očních důlcích)
- Pokrok ve střední části obličeje
- Bi-přepážka na obličej pro rozšíření horní čelisti
- Osteotomie (expanze horní a dolní čelisti)
- Rinoplastika (plastická operace nosu)
- Genioplastika (plastická operace brady nebo tváří)
- Operace očních víček
- Oddělení prstů a / nebo prstů na nohou
- Srdeční chirurgie pro vrozené srdeční vady
Lidé s Apertovým syndromem možná budou muset navštívit speciální lékaře, zejména v dětství, aby zvládli problémy, jako je rozštěp patra a problémy se sluchem. Mohou také těžit ze služeb včasné intervence, jako je logopedie, pracovní terapie a fyzikální terapie, pokud vykazují známky zpoždění vývoje.
Někteří lidé s Apertovým syndromem mají mentální postižení nebo zpoždění, ale mnozí jsou schopni dohnat své vrstevníky.
Zvládání
Mít dítě se speciálními potřebami může být pro každého rodiče zdrcující. Pokud vaše dítě dostane prenatální diagnózu Apertova syndromu, poraďte se se svým lékařem o tom, co můžete očekávat. Genetické poradenství je naprosto doporučeno, protože genetický poradce může nejen vysvětlit příčiny Apertova syndromu, ale také poradit o možnostech mít další děti s Apertovým syndromem. Shromažďujte informace z důvěryhodných zdrojů a pokud je to možné, promluvte si s ostatními rodiči. Ačkoli se diagnóza na první pohled může zdát ohromující a děsivá, můžete se dozvědět, že je zvládnutelnější, než jste původně očekávali.
Apertův syndrom je vzácný stav, ale ve Spojených státech a ve světě je k dispozici více zdrojů a podpůrných skupin. S internetem a sociálními médii je snadnější než kdy jindy spojit se s jinými rodinami a najít podporu. Podívejte se také na kraniofaciální centra ve vašem okolí. Čím více zdrojů budete moci najít a zapojit, tím pohodlněji se budete cítit.
Slovo od Verywell
Diagnóza Apertova syndromu může být pro každého děsivá a obtížná. Není to něco, o čem většina lidí slyšela, a může způsobit významné zdravotní komplikace. Existuje však mnoho zdrojů na pomoc rodinám, aby děti a dospělí s Apertovým syndromem mohli žít a prospívat v dnešním světě.



















-symptoms-and-treatment.jpg)



